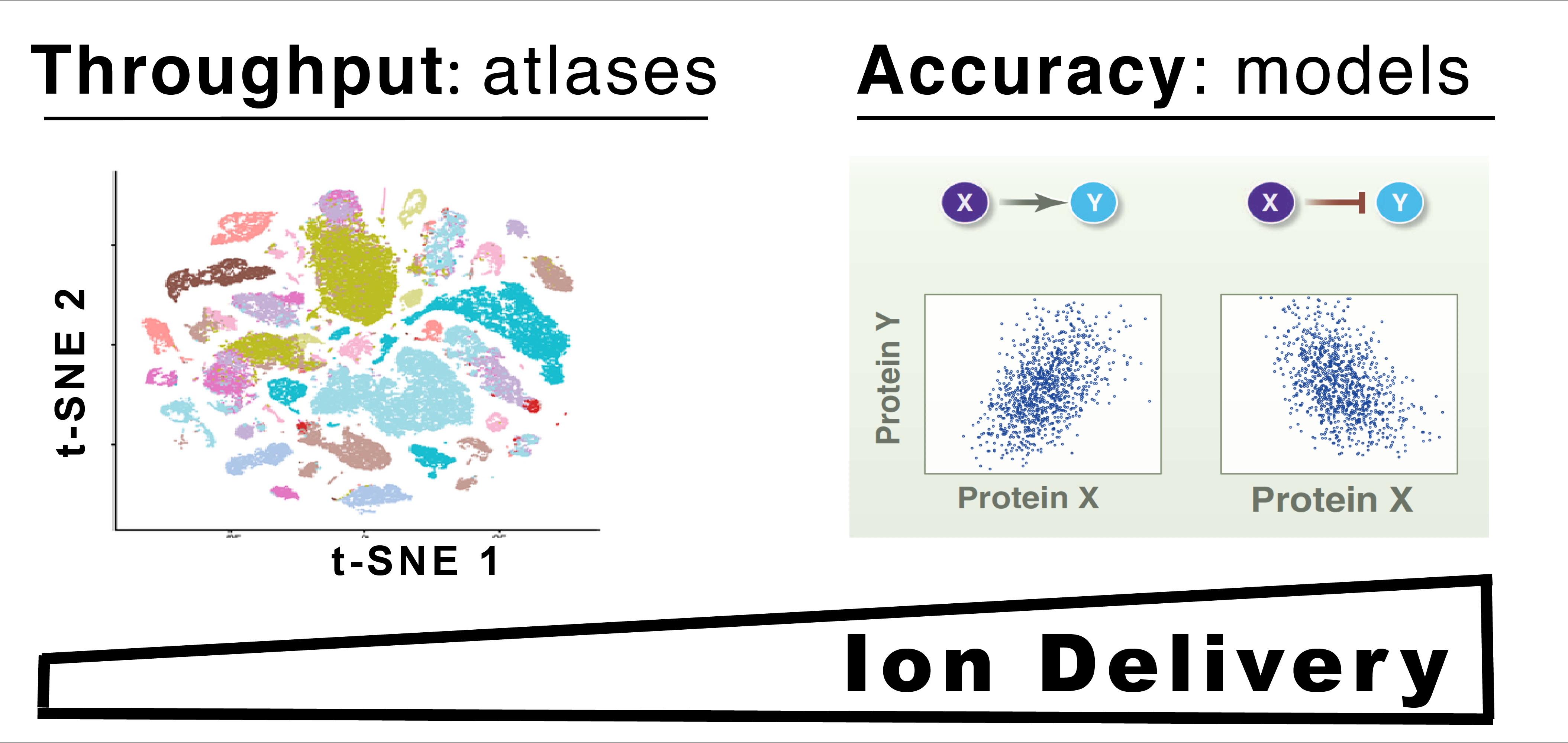
Isobaric carrier optimization
JPR Article bioRxiv Preprint GitHub Code
Table of Contents
Abstract
The isobaric carrier approach, which combines small isobarically-labeled samples with a larger isobarically-labeled carrier sample, is finding diverse applications in ultrasensitive mass-spectrometry analysis of very small samples, such as single cells. To inform the growing use of isobaric carriers, we characterized the trade-offs of using isobaric carriers in controlled experiments with complex human proteomes. The data indicate that isobaric carriers directly enhances peptide sequence identification without simultaneously increasing the number of protein copies sampled from small samples. Furthermore, the results indicate strategies for optimizing the amount of isobaric carrier and analytical parameters, such as ion accumulation time, for different priorities such as improved quantification or increased number of identified proteins. Balancing these trade-offs enables adapting isobaric carrier experiments to different applications, such as quantifying the proteome of limited biopsies or organoids, building single-cell atlases, or modeling protein networks at single-cell resolution. In all cases, the reliability of protein quantification should be estimated and incorporated in all subsequent data analysis. We expect that these guidelines will aid explicit incorporation of the characterized trade-offs in experimental designs and transparent error propagation in the data analysis.
Data from experiments with increasing isobaric carriers
- MassIVE Repository:
DO-MS Reports
These reports contain plots with detailed information for the chromatographic separation (elution peaks), precursor ion intensities, ion accumulation times, apex sampling, number of MS2 scans per experiment and the confidence of the assigned PSMs, and contaminants. This information is provided as distributions across all ions as previously described. For more information, see Data-driven Optimization of Mass-Spectrometry (DO-MS).
- DO-MS Report for the low MS2 AGC target
-
DO-MS Report for the high MS2 AGC target
Guidelines for optimizing isobaric carrier experiments
-
The size of the isobaric carrier should reflect the project priorities. The data reinforce previous suggestions that isobaric carriers that are about 200-fold larger than the small samples provide most of the needed increase in peptide fragments to enhance sequence identification without adverse affects on quantification. Larger carrier sizes can further benefit peptide sequence identification but may result in reduced copy number sampling and noisier data.
-
Evaluate whether the isobaric carrier affects peptide sampling. We recommend estimating whether the carrier levels and AGC target result in reduced accumulation times and sampling of proteins from the small sample. This can be visualized by plotting the distributions of accumulation times and reporter ion intensities. These distributions can be automatically generated by Data-driven Optimization of MS (DO-MS).
-
Estimate the reliability of quantification for each protein. Estimate the sampling error on a per-protein basis as previously demonstrated, benchmark protein quantification or estimate the reliability of quantification based on the consistency of the quantification of peptides originating from the same protein as demonstrated by Franks and colleagues. If the reliability of quantification is limited by counting noise, improved sampling can increase reliability. However, if the reliability is limited by other factors, such as sample preparation, improved copy-number sampling may have limited benefits.
-
Incorporate estimates of reliability in all subsequent analysis. Data points should be weighted based on their reliability, with weights proportional to the reliability. Use error propagation methods to reflect the noise in the final results. For example, correlations between noisy variables can be divided by the corresponding reliability to estimate the fraction of explained variance independent from the noise.
-
Media
About the project
This project on characterizing the isobaric carrier was conducted in the Slavov Laboratory and SCP Center at Northeastern University, and was authored by Harrison Specht and Nikolai Slavov. Learn more about single-cell mass-spectrometry analysis.
This project was supported by funding from the NIH Director’s Award and by an Allen Distinguished Investigator Award from the Paul G. Allen Frontiers Group.