Technical improvements
Decreasing coisolation Apex targetting
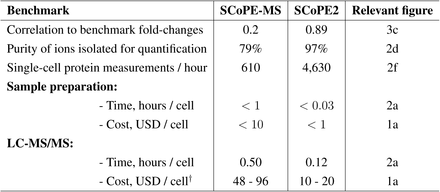
To increase the throughput and quantitative accuracy of single-cell protein analysis by SCoPE-MS, we introduced many technical improvements in both the sample preparation and in the mass-spectrometry analysis. The synergistic effect is to increase quantitative accuracy by 4-fold and the throughput of data acquisition about 8-fold. Below, we outline controlled experiments that illustrate the benefits of individual improvements. To comprehensively compare the mass-spec data at all levels (including chromatographic separation, precursor abundance, ion isolation, spectral purity, and peptide sequence identification), we include the full Data-driven Optimization of MS (DO-MS) reports for each set of experiments.
{kind=link}
Decreasing co-isolation with narrow isolation windows
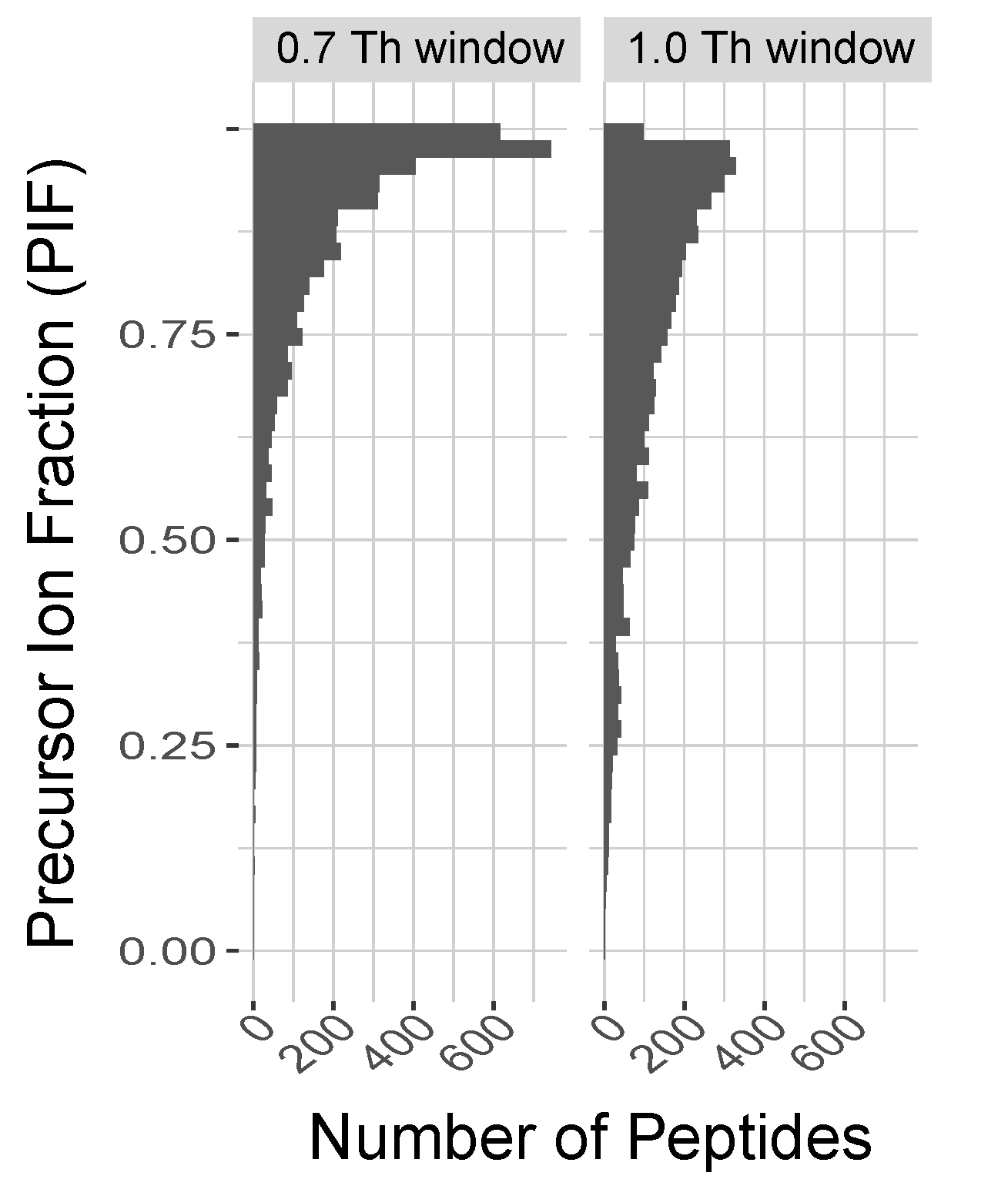
The degree of coisolation (or purity of MS2 spectra) was evaluated as a function of the width of the isolation window. MaxQuant uses the precursor intensity fractions (PIF) to estimate the fraction of the ion intensity originating from the precursor assigned to a peptide spectral match for each MS2 spectrum. The distributions of PIF values for two controlled experiments indicate that the isolation window width used by SCoPE2 (0.7 Th) results in purer spectra compared to the isolation with used with SCoPE-MS. The associated DO-MS report includes dozens of additional plots demonstrating that these experiments were well controlled. It also shows that the wider isolation window of 1 Th results in slightly more identified peptides.
Improving apex targetting
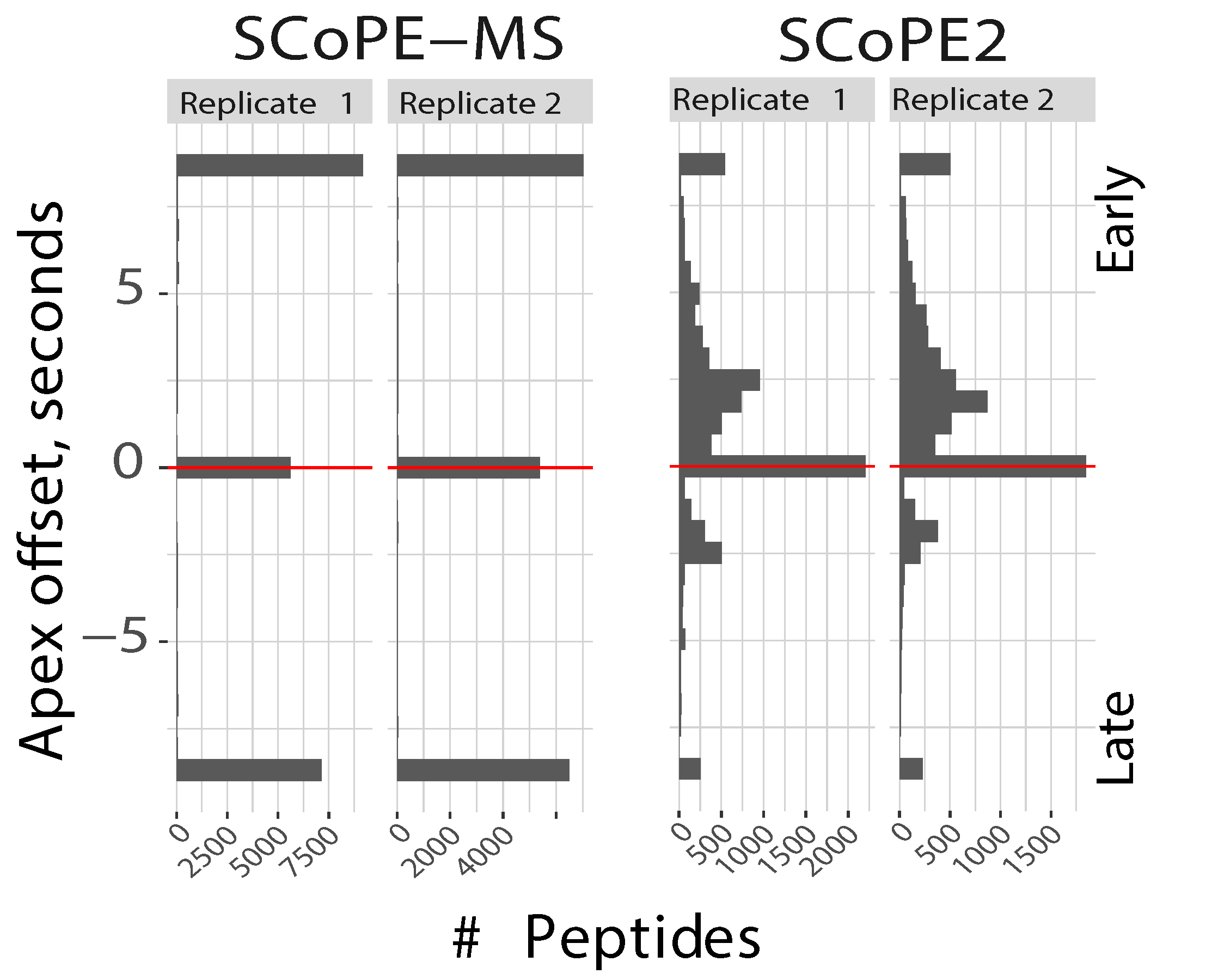
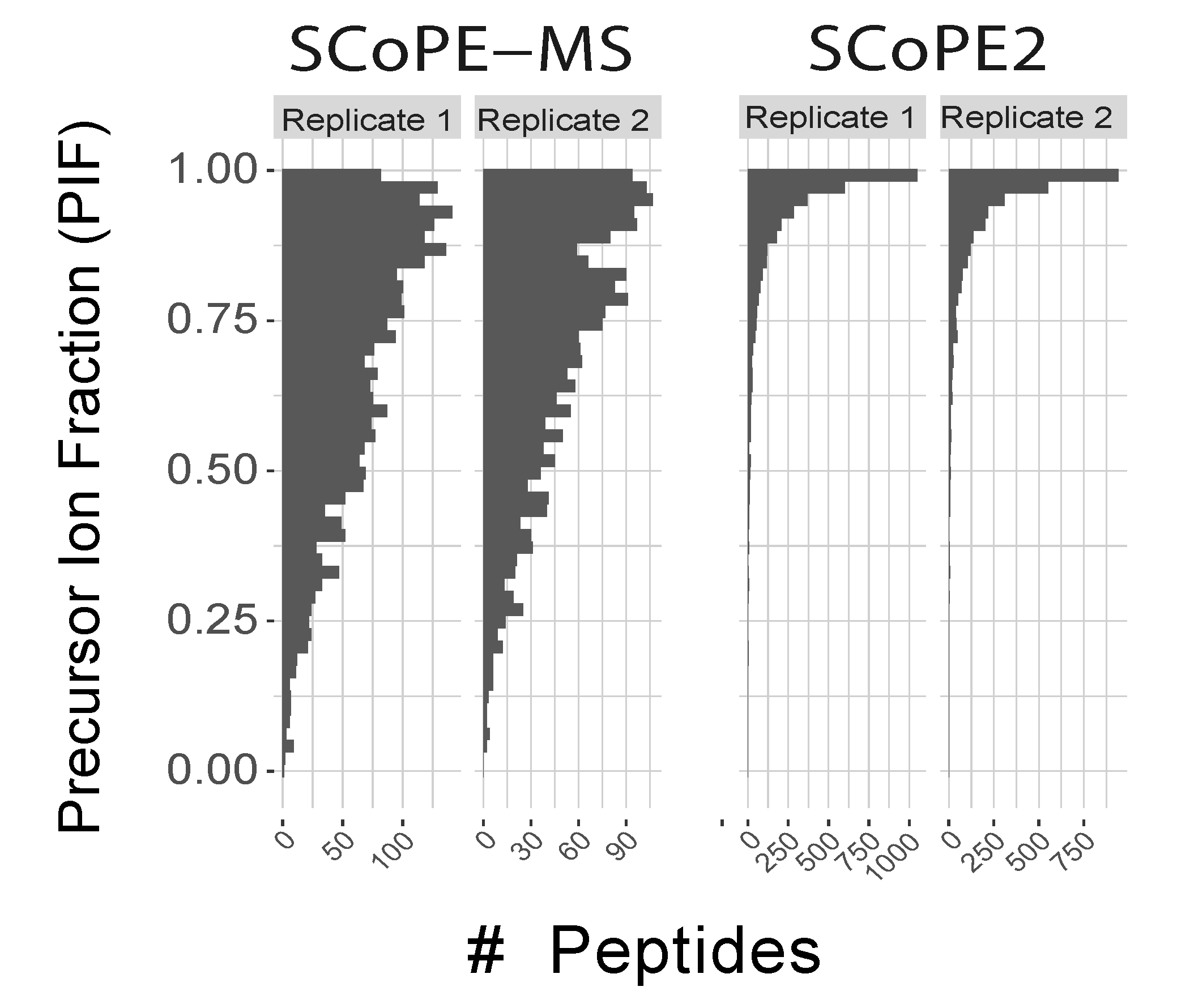
Distributions of apex offsets for data from SCoPE-MS and from SCoPE2. These offsets are computed by MaxQuant as the time between the apex of the elution peak and the time when the ion is sampled for MS2 analysis. The discreetness of the SCoPE-MS distributions is due to the long duty cycles, and thus low time resolution of sampling the elution peaks. The application of DO-MS allowed sampling elution peaks closer to their apecies. This improved apex sampling with SCoPE2 results in much lower coisolation, qantified by the precursor ion fractions (PIF), which MaxQuant computes as estimates for spectral purity, i.e., the fraction of the ion intensity originating from the precursor assigned to a peptide spectral match for each MS2 spectrum.